
Key Points:
- NAD+ declines with age largely due to the activation of CD38, an enzyme activated by the inflammation produced by senescent cells – cells that no longer grow or replicate.
- In a feed-forward cycle, the decline of NAD+ by CD38 induces the spread of more senescent cells and inflammation.
- Halting this cycle could prevent age-related diseases but requires a better understanding of the underlying basis of aging.
Surmounting research places chronic inflammation as a central player of age-related disease. However, inflammation’s role as a middle-man between two other very important underliers of aging may not be as well known: senescence and NAD+ decline. Senescent cells – cells that have stopped growing and dividing – are a major contributor to age-related chronic inflammation. By inducing inflammation, senescent cells begin a cycle that leads to the depletion of NAD+. This cycle spirals out of control as the depletion of NAD+ activates more senescent cells. At the helm of this vicious cycle’s destructiveness is CD38, an enzyme that breaks down NAD+. Understanding how these pieces fit into the aging puzzle can help scientists hone in on intervention points to prevent and treat different age-related diseases.
What Triggers Senescence?
Perhaps contrary to popular belief, scientists think that senescence was coopeted by cells as a mechanism to prevent the spread of genetically damaged cells. For example, since cancer is typically caused by a mutation in our DNA that leads to uncontrollable cell growth and replication, senescence can be activated to stop the propagation of cells with these genetic errors.
There are, however, different types of senescence, based on the tissue and inducer/stressor. Cells can become stressed in many ways, from being pulled or pushed or heated or cooled, but the cell stressor that appears to be at the core of cellular senescence is DNA damage. All sorts of environmental factors, such as UV radiation, toxins, the accumulation of metals, and even chemicals in the food we eat and the air we breathe can impart damage onto our DNA.
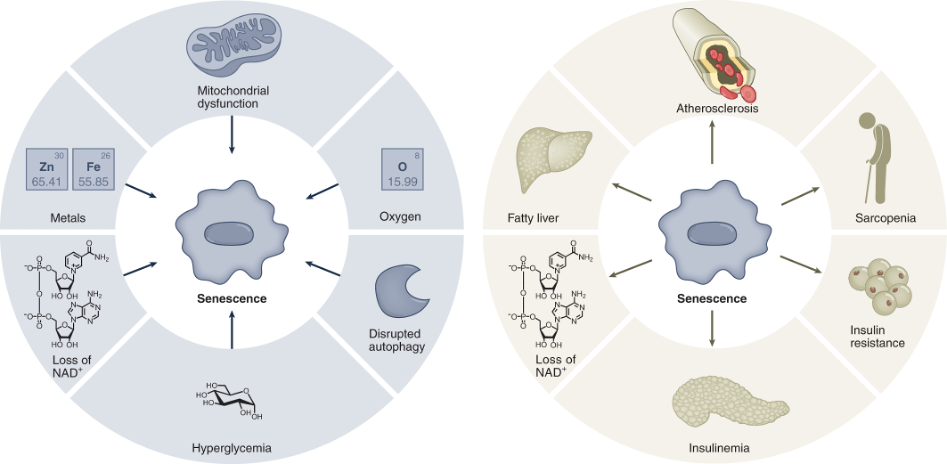
(Wiley and Campisi, 2021 | Nature Metabolism) Drivers of Senescence. Senescent cells can be induced by several stressors, including mitochondrial dysfunction, high oxygen levels, disrupted cellular debris clearance (autophagy), high blood glucose (hyperglycemia), loss of NAD+, and accumulation of metals. In turn, senescence leads to loss of NAD+ and age-related diseases.
Senescence Ignites Inflammation
Once senescent cells are triggered and begin to build up with age, a vicious cycle begins. While senescent cells can be beneficial, playing critical roles in embryonic development, wound healing, and tissue repair, they can become harmful with aging. Senescent cells express molecules that protect them from cell death, so they build up as we grow older. The reason that a senescent cell’s ability to evade self destruction is so detrimental is that they sit at the helm of a persistent inflammatory response by secreting pro-inflammatory signaling molecules.
As senescent cells accumulate with age, more and more inflammation ensues. It could be that the inflammation caused by senescent cells is the root cause of aging. Genetic studies have shown that compared to other factors, senescence is linked to the highest number of age-related diseases. This is likely due to the inflammatory molecules senescent cells secrete.
Inflammation occurs to help fight off pathogens and repair/maintain organs, and is beneficial when turned on transiently. However, when kept on, it becomes chronic and damaging to the organs. For most individuals, aging comes with low-grade chronic inflammation. Inflammation is such a common symptom of aging that it has been termed inflammaging. Senescent cells have been said to be both the cause and consequence of inflammaging.
How Does NAD+ Decline Drive Aging?
Senescent cells and inflammaging are major drivers of aging because they lead to the decline of NAD+. NAD+ serves many cellular functions that help to keep us alive. It is a key molecule involved in mediating the conversion of the food we eat into cellular energy. Proteins use NAD+ to regulate an array of vital cellular processes. The importance of NAD+ is emphasized by its role in aging. NAD+ declines with age, and its depletion is associated with nearly all age-related diseases.
This occurs because without NAD+, cells can no longer function properly. Once the cell is unable to efficiently produce cellular energy (ATP) from the food we eat, it’s machinery starts to break down. One of the major indicators of cell metabolic failure is mitochondrial dysfunction, as mitochondria are the cell organelles responsible for producing cellular energy. As cells malfunction, so do organs, leading to the diseases we see with aging.
Several factors contribute to the drop in NAD+ that occurs with aging. These factors include a decrease in enzymes that help synthesize NAD+ and an increase in several proteins that degrade NAD+. Among these factors, CD38 appears to have the most significant contribution to the decline of NAD+ during aging. CD38 effectively closes the cycle, as it is the primary mediator between inflammation and NAD+ depletion.
What is CD38?
In response to pathogens, the immune system attempts to eliminate foreign particles from the body. This immune response includes the activation of CD38, which helps stop infections and tumor growth. However, like with many biological processes, too much of a good thing can go wrong. Too much CD38 activity can disrupt the integrity of organs by breaking down NAD+.
CD38 seems to break down NAD+ to defend against pathogens. CD38 breaks down other molecules, but the breakdown of NAD+ accounts for about 90% of its enzymatic activity. Some scientists, like Hogan and colleagues, have proposed that the purpose of CD38 is to break down NAD+. There are several clues to support this hypothesis.
The first clue is that CD38 is expressed mostly on the surface of immune cells where it faces outward, instead of facing towards the inside of the cell where most of the NAD+ is located. The second clue is that, in addition to NAD+, CD38 breaks down the precursors of NAD+; NMN (nicotinamide mononucleotide) and NR (nicotinamide riboside), which are needed to synthesize NAD+.
Another clue is that some species of bacteria, such as H. influenza, cannot make their own NAD+, which they need to survive. Many bacteria rely on the uptake of NAD+ or its precursors for growth. Therefore, by depriving the extracellular environment of NAD+ and its precursors, CD38 can effectively kill bacterial pathogens.
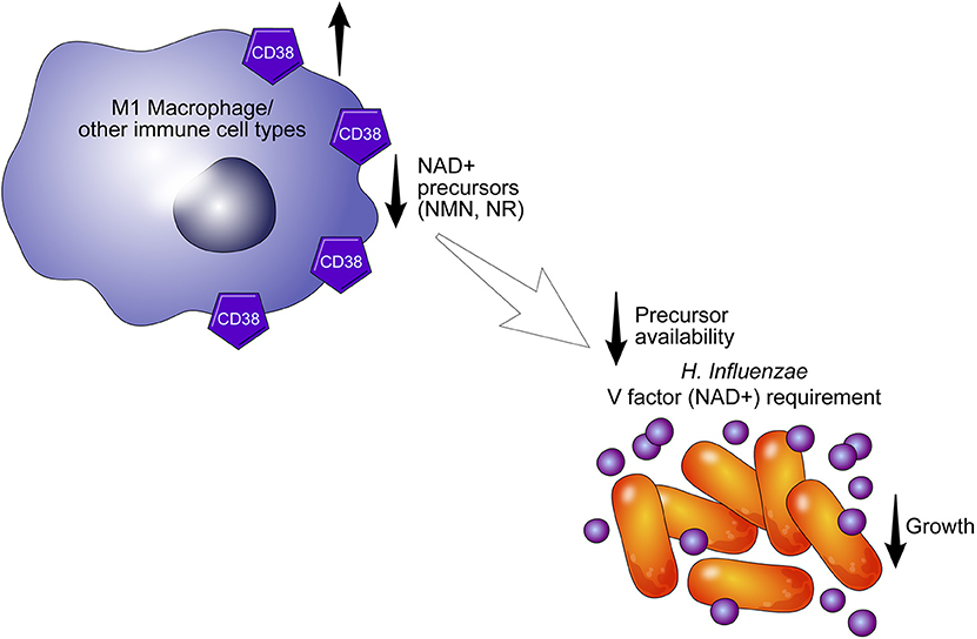
(Hogan et al., 2019 | Frontiers in Immunology) CD38 Immune Cells Starve Bacteria. The CD38 enzyme on the surface of an immune cell (macrophage, or other) breaks down NAD+ and its precursors to decrease the growth of H. Influenzae.
Whatever its true nature, CD38 seems to have a profound effect on aging by consuming NAD+. Overall, the increased activation of CD38 that occurs with aging contributes largely to the breakdown and decline of NAD+, leading to age-related disease. NAD+ dysregulation and increased CD38 are shared by many diseases.
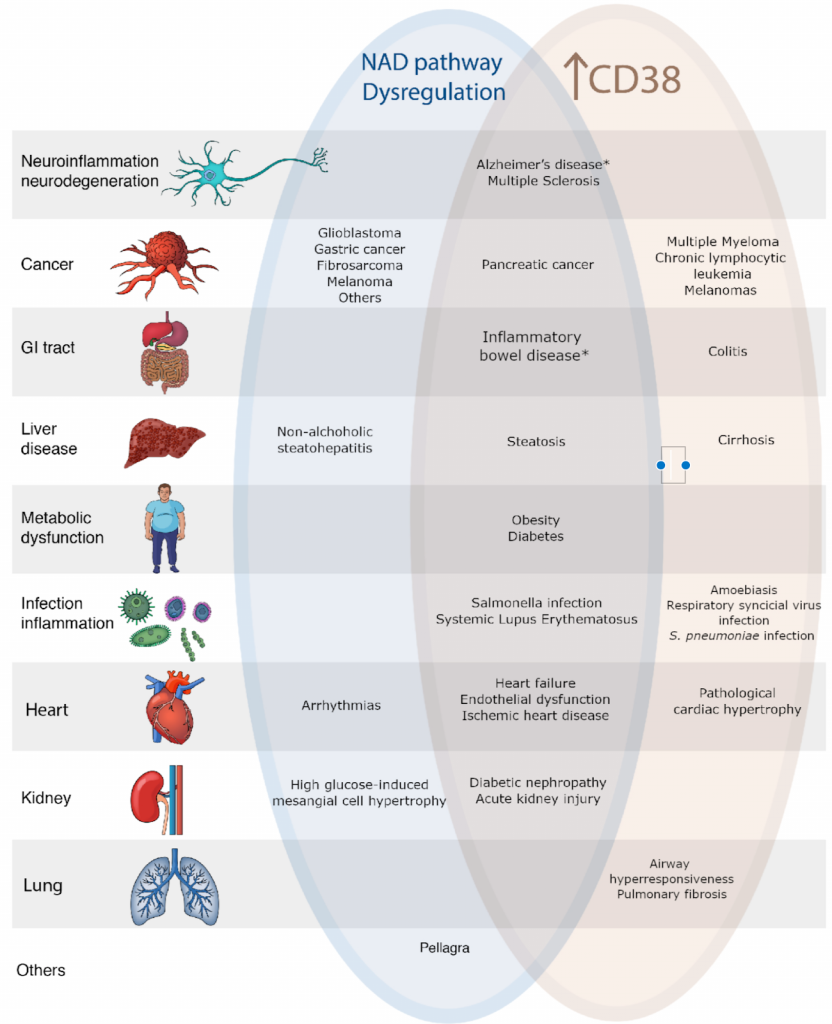
(Zeidler et al., 2022 | Am J Physiol Cell Physiol) NAD+ Depletion and CD38 Contribute to Disease. CD38 breaks down NAD+. With aging, levels of NAD+ decline while levels of CD38 increase. Both are associated with some of the same diseases.
A Vicious Cycle
Overall, the inflammatory molecules that senescent cells secrete can activate CD38, primarily on immune cells. Inflammation and CD38 activation are usually temporary, but when senescent cells accumulate with age, they can lead to chronic activation of CD38 through inflammation, thereby contributing to the age-related decline of NAD+. Interestingly, NAD+ loss is also a driver of senescence. And here we have a vicious cycle. As NAD+ is depleted, more senescent cells are induced, activating CD38 to further deplete NAD+. Without intervention, this cycle seems to exacerbate the aging process.
Ending the Cycle
If senescent cells are the root of chronic inflammation and NAD+ depletion, which underlie almost every age-related disease, why not just get rid of them? While most senescent cells are protected from normal cell death, they can be killed by drugs called senolytics. While senolytics seem promising, as they mitigate age-related disease, the diversity of senescence makes it complicated. For example, brain cells can become senescent, but we can’t make more brain cells like we can skin cells. Therefore, it might not be a good idea to kill our precious brain cells with senolytics. Instead of eliminating senescent cells, it may instead be better to reverse the senescent state. We are not yet at the point where this can be done with a drug, but this may become possible in the future.
Instead of eliminating or reverting senescent cells, CD38 inhibitors or NAD+ boosters can also be utilized to possibly deter the negative effects of aging. These specific targets bypass the complexity of senescent state variation, which we may better understand in the coming years. Indeed, there are still many questions that need to be answered before understanding the true nature of senescence.
One question that has yet to be answered is why senescent cells are not eliminated or reversed naturally. The life expectancy charts have shot up in recent years, so it’s possible that the process of natural selection hasn’t had a chance to catch up. Life expectancy has increased largely due to technological advances that do not address underlying biological processes, like the induction of senescent cells. Now it seems that we can overstep evolution once more as we learn more about senescence. We can even accelerate aspects of evolution by finding the genes that make us live longer. We know there is a gene related to eliminating senescent cells, and the individuals with this gene tend to live longer.
 Comments
Comments