
Degeneration of axons, which transmit signals from one neuron to another, occurs in neurological diseases. A molecule called nicotinamide adenine dinucleotide (NAD+) has a critical role in axon maintenance through unclear influences on a protein called Sarm1 that when activated causes neuron degeneration, the progressive atrophy and loss neuron function.
Scientists from Peking University in Beijing, China, published an article in Nature where they found NAD+ binds to a region on human Sarm1 that inhibits its pro-neurodegenerative activity. Furthermore, disrupting the Sarm1 binding region to NAD+ stimulated the protein and facilitated axon degeneration in neurons from mice. These findings suggest a mechanism whereby NAD+ mediates Sarm1 inhibition and the degeneration of neurons.
NAD+ is a crucial molecule for energy production and maintaining health in cells. Levels of cell NAD+ diminish with age, which scientists have linked to certain age-related neurodegenerative diseases, like Alzheimer’s disease. How NAD+ levels in cells influence neuron degeneration has not been fully defined.
Sarm1 activation promotes the molecular breakdown of NAD+ and induces degeneration of neurons. It has three protein regions known as ARM, TIR, and SAM, and the interactions between these domains influence its neurodegenerative activity.
In the study, the researchers used a high resolution imaging technique called electron microscopy to look at the fine, molecular details of Sarm1. They found that the ARM region interacts with the TIR region and that NAD+ binding to the ARM region inhibits the activity of the TIR domain along with Sarm1’s activation that promotes neuron degeneration.
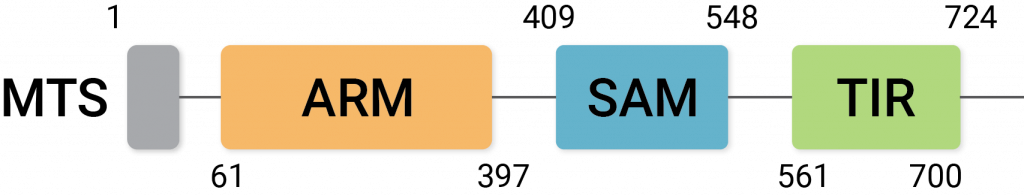
(Jiang et al., 2020 | Nature) An illustration of the three domains of Sarm1: the ARM, SAM, and TIR domains.
Mutations inhibiting the ARM region and TIR region interaction induced extensive axonal degradation. These findings showed that the ARM domain interacts with the TIR domain to inhibit Sarm1’s neurodegenerative activity.
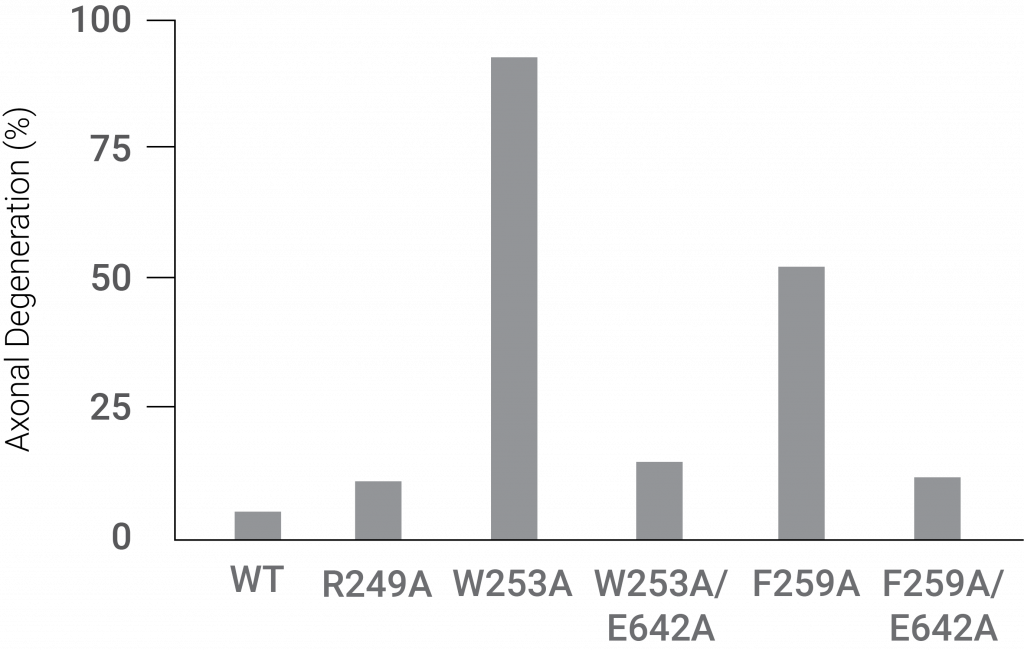
(Jiang et al., 2020 | Nature) Two mutations (W253A and F259A) in regions of the ARM domain of Sarm1 that disrupt its interaction with the TIR domain facilitate dramatic axonal degeneration.
Moreover, Sarm1 was activated by blocking access to its NAD+ binding site along with the interaction between the ARM and TIR domains, which led to axon degeneration. These findings suggest that NAD+-mediated Sarm1 self-inhibition causes neuron degeneration.
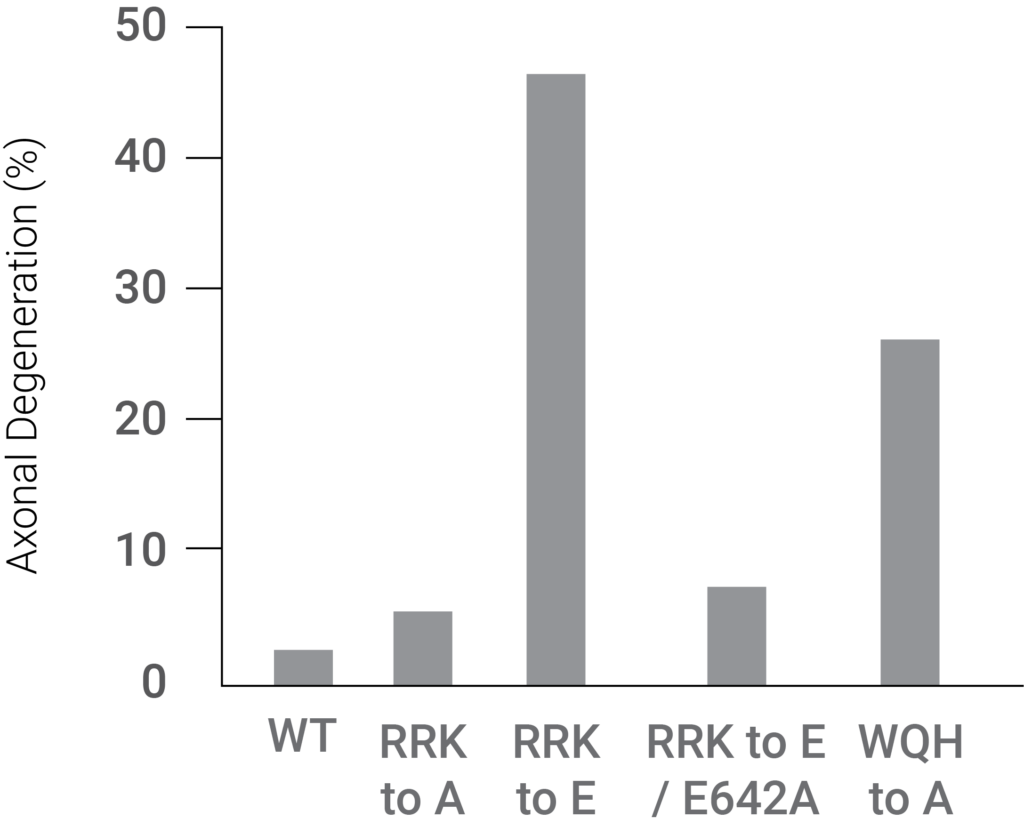
(Jiang et al., 2020 | Nature) A mutation in the Sarm1 ARM domain that disrupts NAD+ binding facilitates dramatically increased axon degeneration. The disruptive mutation, referred to as RRK to E, resulted in significant axonal degeneration. The figure shows the drastic axonal degeneration elevation associated with this mutation.
“Of particular interest, we discovered NAD+ as the novel ligand to Sarm1 ARM,” stated the investigators in their study. “The NAD+ binding could facilitate the protein’s self-inhibition, as the mutations impairing NAD+ binding produced the constitutively-active Sarm1. Notably, this NAD+-mediated self-inhibition mechanism is supported by a separate study,” they said in reference to the neurodegenerative-causing activities of Sarm1 with disrupted NAD+ binding. Importantly, these findings could also have clinical implications.
“The newly-revealed NAD+-binding site may become a promising drug target,” they said. “Small-molecule analogs to NAD+ would block the Sarm1 activation…Such compounds could have broad implications for the treatment of different neurodegenerative diseases.”
 Comments
Comments